This is a directory to scientific software from our lab that is freely available to the academic community. Most of our developments are concerned with postprocessing of cryo-EM maps and atomic model refinement using cryo-EM data. Please visit the individual pages for more details if you are interested. You can also find a collection of potentially useful jiffys and tools for cryo-EM model building.
All projects are hosted by our group respository on Github or TU Delft Gitlab.
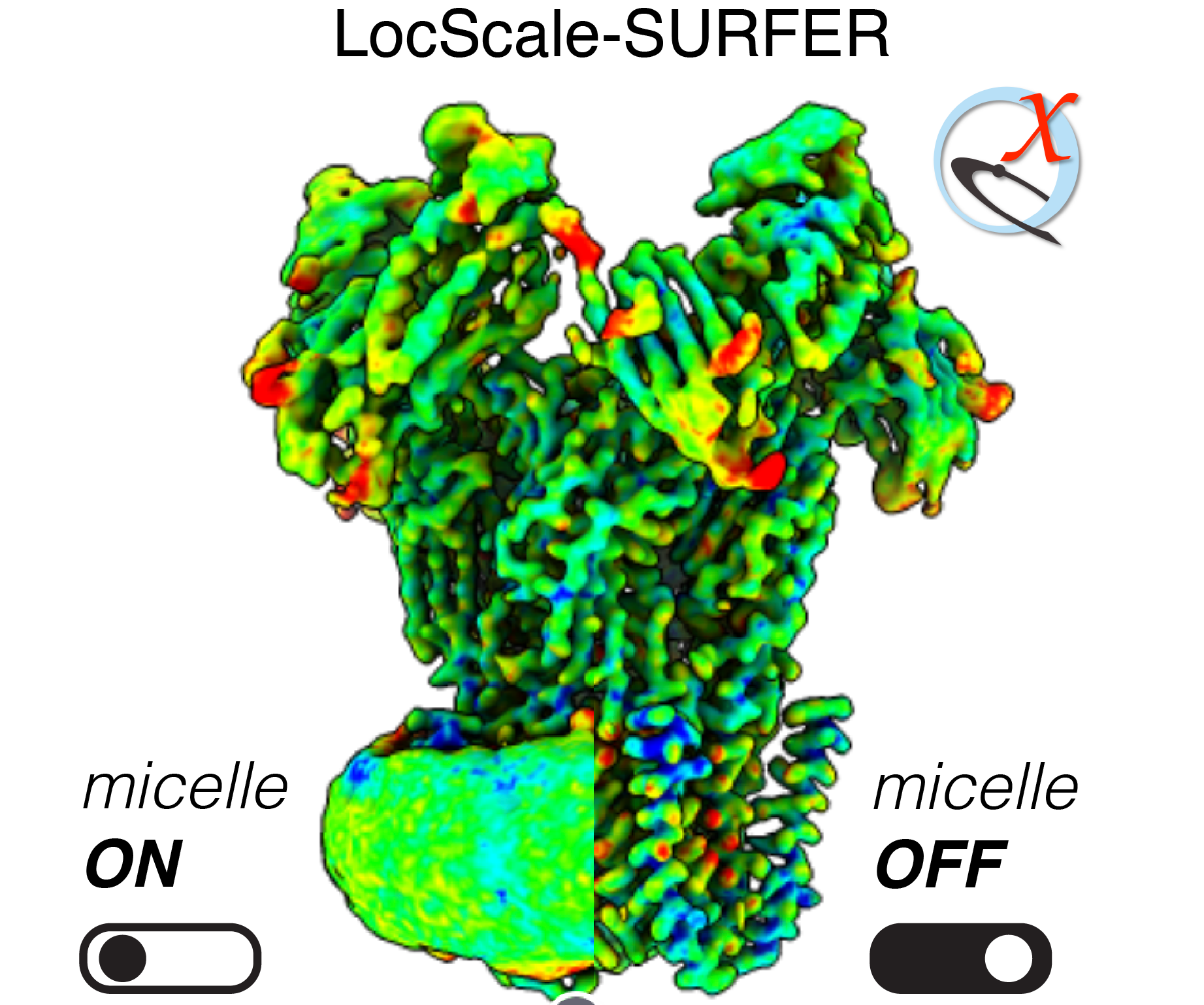 LocScale-SURFER is a ChimeraX extension bundle for enhancing visualisation and interpretation of LocScale-optimised maps containing contextual structure. It currently supports the detection and targeted segmentation/removal of micellar/membranous structures, but may be extended to other contextual structures in the future.
LocScale-SURFER is a ChimeraX extension bundle for enhancing visualisation and interpretation of LocScale-optimised maps containing contextual structure. It currently supports the detection and targeted segmentation/removal of micellar/membranous structures, but may be extended to other contextual structures in the future.
ColabScale: LocScale-SURFER is also integrated in ColabScale, which provides easy-to-use access for all LocScale-2.0 workflows via Google Colab.
Availability:
LocScale-SURFER is freely available under the BSD licence. For more information, tutorials as well as installation instructions please visit the documentation pages. LocScale-SURFER can be installed in existing ChimeraX instances via the ChimeraX Toolshed.
If LocScale-SURFER and/or ColabScale are useful for you work please consider citing the references below.
Bharadwaj et al. BioRxiv 2026.02.25.707988 (2026) | doi |
![]()
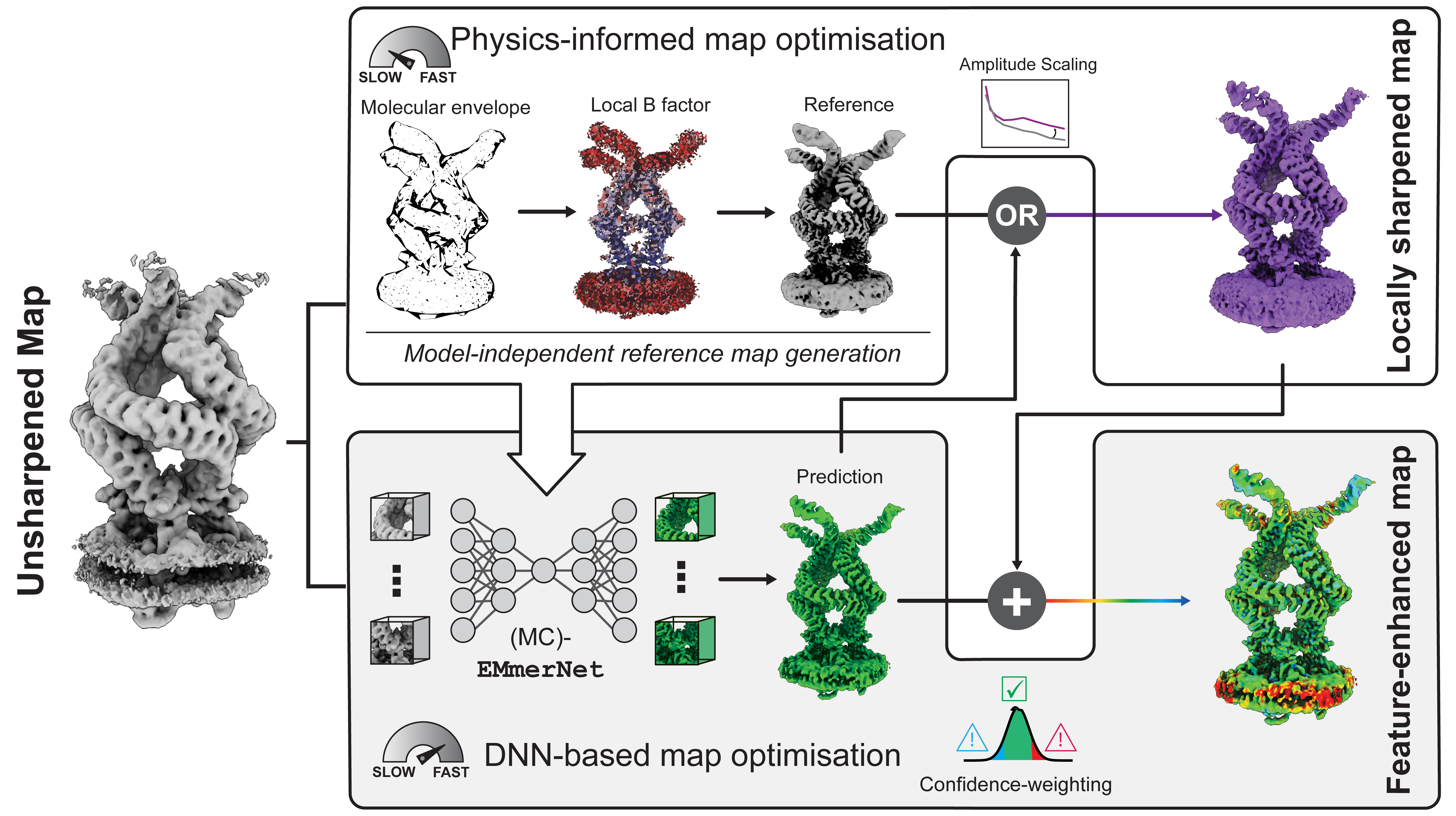 LocScale 2.0 is an automated program for physics-informed local sharpening and/or density modification of cryo-EM maps with the aim to improve their interpretability. It utilises general properties inherent to electron scattering from biological macromolecules to restrain the sharpening filter. These can either inferred directly from the experimental density map, or be provided from an existing atomic model.
LocScale 2.0 is an automated program for physics-informed local sharpening and/or density modification of cryo-EM maps with the aim to improve their interpretability. It utilises general properties inherent to electron scattering from biological macromolecules to restrain the sharpening filter. These can either inferred directly from the experimental density map, or be provided from an existing atomic model.
ColabScale: ColabScale provides easy-to-use access for all LocScale-2.0 workflows via Google Colab.
Availability:
LocScale-2.0 is freely available under the BSD licence. For more information, tutorials as well as installation instructions please visit the documentation pages. LocScale-2.0 is also distributed together with the CCP-EM Doppio platform and instructions for using LocScale-2.0 in Doppio can be found here.
If LocScale-2.0 and/or ColabScale are useful for you work please consider citing the references below. If you utilise the CCP-EM implementation of LocScale2.0 please also credit the CCP-EM project.
Bharadwaj et al. BioRxiv 2025.09.11.674726 (2025) | doi |
Bharadwaj et al. Faraday Discuss. 240:168-183 (2022) | doi |
![]()
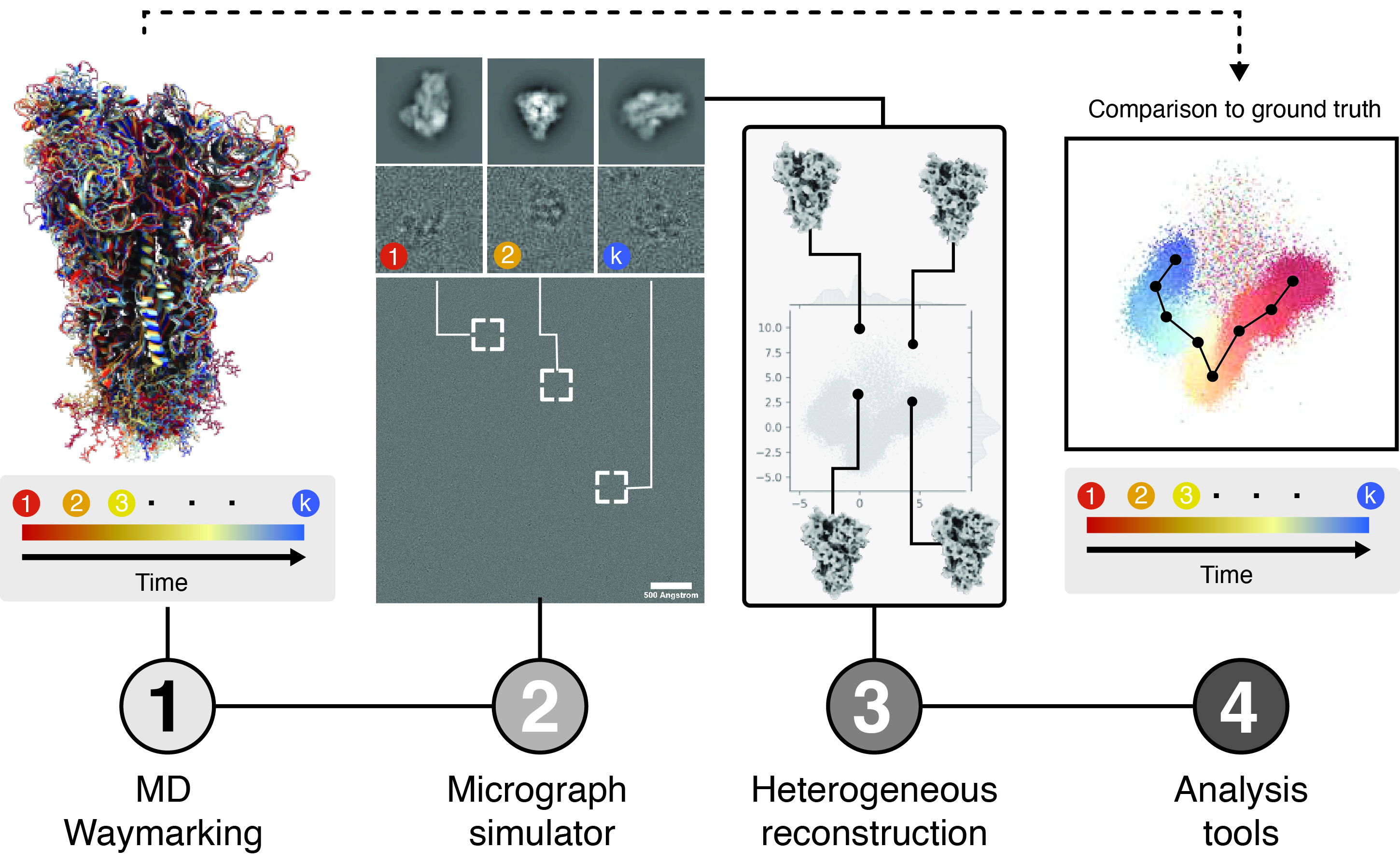 Roodmus is computational toolkit that allows sourcing conformational heterogeneity of biological macromolecules from atomistic molecular dynamics simulations to generate realistic, synthetic cryo-EM datasets, and parsing information about the exact pose and conformation of each particle along the entire reconstruction workflow. This allows the results of heterogeneous reconstruction algorithms and the effect of experimental factors such as radiation damage, preferred orientations or electron optical aberrations on their outcome, to be systematically investigated by comparison to ground truth information.
Roodmus is computational toolkit that allows sourcing conformational heterogeneity of biological macromolecules from atomistic molecular dynamics simulations to generate realistic, synthetic cryo-EM datasets, and parsing information about the exact pose and conformation of each particle along the entire reconstruction workflow. This allows the results of heterogeneous reconstruction algorithms and the effect of experimental factors such as radiation damage, preferred orientations or electron optical aberrations on their outcome, to be systematically investigated by comparison to ground truth information.
Availability:
roodmus is freely available under the GPL licence. For more information as well as download/installation instructions please visit the repository. A detailed description of use, best practices and analysis tools are available on the README and scripts directory.
roodmus was developed in collaboration with Joel Greer and Tom Burnely at CCP-EM.
If roodmus is useful for you work please consider citing the original reference. If you utilize the CCP-EM implementation of roodmus please also credit the CCP-EM project.
Joosten, Greer et al., IUCrJ 11:951-965 (2024) | doi |
![]()
![]() LocScale is a reference-based local amplitude scaling tool using prior model information to improve contrast of cryo-EM density maps. It can be helpful in the common case of resolution variation in the 3D reconstruction and it can be used as an alternative to other commonly applied map sharpening methods.
LocScale is a reference-based local amplitude scaling tool using prior model information to improve contrast of cryo-EM density maps. It can be helpful in the common case of resolution variation in the 3D reconstruction and it can be used as an alternative to other commonly applied map sharpening methods.
Availability:
LocScale is freely available under the BSD licence. For more information as well as download/installation instructions please visit the Gitlab repository. Tutorial files and a detailed description of use and best practices are available on the Wiki pages.
NEW:
An accelerated and GUI-supported version of LocScale has now been integrated into the CCP-EM suite and is distributed with the latest release v1.2.0.
If LocScale is useful for you work please consider citing the original reference. If you utilise the CCP-EM implementation of LocScale please also credit the CCP-EM project.
Jakobi et al., eLife 6:e27131 (2017) | doi |
Burnley et al., Acta Cryst. D73:469-477 (2017) | doi |
![]()
![]()
PySerialEM is a small Python library to read/write and manipulate SerialEM navigator (.nav) files, or to process (e.g. montage) SerialEM grid maps.
Availability:
PySerialEM is freely available under the BSD licence. General instructions can be found here.
NEW: See here for Stef’s demo on grid montaging.
![]()
rsref is a scripted modular workflow for the refinement of atomic models against high-resolution cryo-EM density maps. It is a not a standalone refinement software, but rather provides a set of tools for model and/or map manipulation, refinement protocols, the analysis of the refinement cycles and validation of the resulting coordinate models.
rsref makes use of the libraries from the cctbx project.
Availability:
rsref is an ongoing development and not officially released. If you are fine with limited support, please see the Wiki pages for dowload and usage instructions and tutorial.
If rsref is useful for you work please acknowledge the cctbx project (doi).
Primary references:
Nature 528:231-236 (2015) | doi |
FEBS J 283:2811–2819 (2016) | doi |
Kavli Institute of Nanoscience
Department of Bionanosciene
Delft University of Technology
Van der Maasweg 9, 2629 HZ Delft